內(nèi)皮細(xì)胞(endothelial cell,EC)遍布血管,是氧氣和營養(yǎng)物質(zhì)輸送、免疫細(xì)胞運(yùn)輸以及清除遠(yuǎn)組織廢物的重要管道。目前認(rèn)為內(nèi)皮細(xì)胞(ECs)代謝是促進(jìn)血管生成的重要因素,ECs不同的代謝途徑,如糖酵解、脂肪酸氧化和谷氨酰胺代謝,在血管形成過程中發(fā)揮了不同的重要作用。研究發(fā)現(xiàn),糖酵解過程中的一個(gè)關(guān)鍵限速酶——磷酸果糖激酶-2/果糖-2,6-二磷酸酶3(phosphofructokinase-2/fructose-2,6-bisphosphatase 3,PFKFB3)具有較強(qiáng)的激酶活性,若抑制其活性可以顯著減少糖酵解,從而抑制病理性血管的生成。ECs還通過血管分泌信號傳導(dǎo)促進(jìn)組織穩(wěn)態(tài)和再生,然而,EC是否參與代謝性血管內(nèi)分泌串?dāng)_以控制局部缺血誘導(dǎo)的肌肉再生尚不清楚。
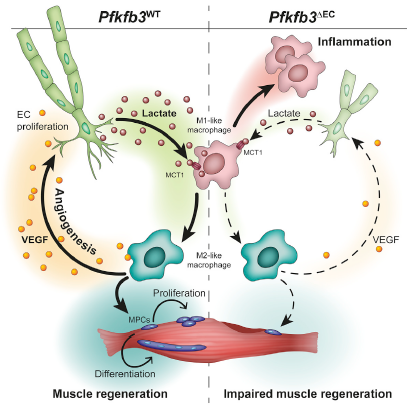
近期,蘇黎世聯(lián)邦理工學(xué)院的 Katrien De Bock團(tuán)隊(duì)(第1作者為張靜)在Cell Metabolism雜志上發(fā)表了題為“Endothelial Lactate Controls Muscle Regeneration from Ischemia by Inducing M2-like Macrophage Polarization”的文章,揭示了內(nèi)皮細(xì)胞通過分泌乳酸介導(dǎo)巨噬細(xì)胞極化來促進(jìn)血管重生和肌肉再生,證明了在肌肉內(nèi)存在第二種乳酸穿梭機(jī)制,即從內(nèi)皮到巨噬細(xì)胞。

巨噬細(xì)胞在骨骼肌再生中起關(guān)鍵作用。研究人員發(fā)現(xiàn),糖酵解調(diào)節(jié)因子pfkfb3在內(nèi)皮細(xì)胞(EC)的特異性缺失可以緩解缺血性后肢受損的血管生成和肌肉再生能力。這是由于巨噬細(xì)胞促血管生成和促再生的M2型表型的能力降低引起的。為了剖析內(nèi)皮細(xì)胞PFKFB3特異性敲除小鼠中M2型極化受損與缺血后肌肉修復(fù)受損的功能相關(guān)性,后肢缺血3天后,研究人員將未極化的骨髓來源巨噬細(xì)胞或M2型巨噬細(xì)胞注射到小鼠后肢,發(fā)現(xiàn)M2型巨噬細(xì)胞顯著促進(jìn)內(nèi)皮細(xì)胞PFKFB3特異性敲除小鼠肌肉修復(fù),但并未*達(dá)到對照組水平。說明 內(nèi)皮細(xì)胞PFKFB3敲除后對缺血后肌肉組織修復(fù)的影響是部分通過對M2型巨噬細(xì)胞極化調(diào)控發(fā)揮作用。
為了研究EC是否利用血管分泌機(jī)制來影響巨噬細(xì)胞極化,研究人員分離了野生型小鼠內(nèi)皮細(xì)胞和PFKFB3敲除小鼠內(nèi)皮細(xì)胞與BMDMs共培養(yǎng),發(fā)現(xiàn)野生型小鼠內(nèi)皮細(xì)胞顯著促進(jìn)BMDMs分化成M2型巨噬細(xì)胞。有趣的是,用mECs-CM(條件性培養(yǎng)基)進(jìn)行的BMDM刺激不能*概括經(jīng)典的IL-4介導(dǎo)的M2極化。從mEC的CM中清除代謝物會減弱其誘導(dǎo)M2型極化的能力。通過對正常內(nèi)皮細(xì)胞和PFKFB3敲除內(nèi)皮細(xì)胞來源培養(yǎng)基的細(xì)胞因子(cytokine)和代謝產(chǎn)物分析發(fā)現(xiàn)內(nèi)皮細(xì)胞敲除PFKFB3后,細(xì)胞因子無顯著變化,而乳酸分泌顯著下降。同時(shí)在補(bǔ)充生理濃度乳酸后,PFKFB3敲除內(nèi)皮細(xì)胞來源培養(yǎng)基促進(jìn)M2型巨噬細(xì)胞極化能力增強(qiáng)。值得注意的是, 乳酸控制巨噬細(xì)胞極化的能力需要條件培養(yǎng)基,這表明功能性極化需要其他細(xì)胞因子或代謝物的存在。
研究人員進(jìn)一步收集了分化后巨噬細(xì)胞培養(yǎng)基,發(fā)現(xiàn)PFKFB3敲除內(nèi)皮誘導(dǎo)的巨噬細(xì)胞培養(yǎng)基分泌更多的VEGF。通過促進(jìn)M2型巨噬細(xì)胞極化(通過移植M2型巨噬細(xì)胞轉(zhuǎn)移或增加乳酸水平)來恢復(fù)內(nèi)皮細(xì)胞PFKFB3特異性敲除小鼠肌肉中的VEGF水平,可增加血管密度,盡管后者在施用乳酸后未能達(dá)到野生型小鼠的水平。這表明觀察到的血管生成缺陷(至少在缺血性肌肉中)是PFKFB3對內(nèi)皮遷移/增殖的直接抑制作用以及肌肉微環(huán)境對血管生成刺激減少的組合。
單羧酸鹽轉(zhuǎn)運(yùn)蛋白1(MCT1)是乳酸的重要運(yùn)載體,為了研究內(nèi)皮細(xì)胞來源乳酸促進(jìn)巨噬細(xì)胞極化的分子機(jī)制,研究人員用巨噬細(xì)胞MCT1特異性敲除小鼠(賽業(yè)生物構(gòu)建)進(jìn)行了更深一步的研究。發(fā)現(xiàn)在下肢缺血條件下,巨噬細(xì)胞MCT1敲除抑制M2型巨噬細(xì)胞的分化,同時(shí)降低了肌肉組織VEGF的分泌。病理學(xué)分析發(fā)現(xiàn)MCT1敲除抑制血管新生和肌肉組織再生。放射性標(biāo)記實(shí)驗(yàn)發(fā)現(xiàn)巨噬細(xì)胞以MCT1作為乳酸運(yùn)載體將內(nèi)皮來源乳酸轉(zhuǎn)運(yùn)到細(xì)胞內(nèi),并作為底物參與到巨噬細(xì)胞氧化代謝。在低氧和營養(yǎng)不足的條件下為巨噬細(xì)胞發(fā)揮功能提供能量。
總之,該研究證明了內(nèi)皮細(xì)胞利用其*的代謝特征在缺血期間以依賴于乳酸穿梭至巨噬細(xì)胞的血管分泌方式調(diào)控肌肉再生。乳酸介導(dǎo)的巨噬細(xì)胞極化促進(jìn)血管生成和肌肉再生。因此,EC特異性pfkfb3的丟失會降低肌肉乳酸水平并損傷缺血后肌肉的修復(fù)。代謝性血管內(nèi)分泌信號提供了一種新型機(jī)制,通過該機(jī)制,EC可以促進(jìn)組織穩(wěn)態(tài)和再生。
原文檢索:
Endothelial Lactate Controls Muscle Regeneration from Ischemia by Inducing M2-like Macrophage Polarization.
DOI: 10.1016/j.cmet.2020.05.004.

掃一掃,關(guān)注我們
